
Seien Sie am 30. April dabei: Vorstellung von Parasoft C/C++test CT für kontinuierliche Tests und Compliance-Exzellenz | Registrierung
Zum Abschnitt springen
Navigieren in den FDA-Validierungsleitlinien für die Cybersicherheit medizinischer Geräte

13. November 2023
13 min lesen
Da die FDA weitere Anforderungen an ihre Cybersicherheitsvalidierung und -standards stellt, müssen Hersteller medizinischer Software statische Analysen einführen, um sicherzustellen, dass ihre Software diese neuen Sicherheitsstandards erfüllt. Lesen Sie weiter, um zu erfahren, wie Sie statische Analysen implementieren, um diese Sicherheitsanforderungen zu erfüllen.
Zum Abschnitt springen
Zum Abschnitt springen
Da die FDA ihre Richtlinien zur Softwarevalidierung um weitere Cybersicherheitsanforderungen erweitert, können Hersteller medizinischer Geräte auf die statische Analyse zurückgreifen, die effektivste Methode, um Sicherheitsbedenken auszuräumen und vorhersehbare Software bereitzustellen.
Hersteller medizinischer Geräte konzentrieren sich aus zwei Hauptgründen weiterhin auf die Verbesserung von Softwareentwicklungsprozessen.
- Bewältigen Sie wachsende Sicherheitsbedrohungen.
- Erfüllen Sie die immer präziser werdenden Anforderungen der FDA.
Verstehen der neuen FDA-Cybersicherheitsleitlinie
Die neuen Leitlinien der FDA zur Cybersicherheit für medizinische Geräte wurden im September 2023 veröffentlicht und enthalten umfassende Empfehlungen für die Branche zum Design, zur Kennzeichnung und zur Dokumentation von Cybersicherheitsgeräten, die in Anträge vor der Markteinführung von Geräten mit Cybersicherheitsrisiko aufgenommen werden sollen.
Der Leitfaden soll Herstellern dabei helfen, Cybersicherheitsrisiken während des gesamten Lebenszyklus eines Medizinprodukts zu erkennen und zu mindern, vom Design und der Entwicklung bis hin zur Herstellung, Wartung und Überwachung nach dem Inverkehrbringen. Darüber hinaus werden Empfehlungen für die Kommunikation von Cybersicherheitsrisiken an Gesundheitsdienstleister und Patienten gegeben.
Die neuen Leitlinien bauen auf den früheren Leitliniendokumenten der FDA zur Cybersicherheit auf, enthalten aber auch eine Reihe neuer und aktualisierter Empfehlungen, wie zum Beispiel:
- Eine Anforderung an Hersteller, ein umfassendes Cybersicherheits-Risikomanagementprogramm zu implementieren.
- Eine Anforderung für Hersteller, eine Software-Stückliste (SBOM) für ihre Geräte einzureichen.
- Eine Anforderung an Hersteller, Cybersicherheitspläne für den Post-Market-Support zu entwickeln und umzusetzen.
- Empfehlungen zur Kommunikation von Cybersicherheitsrisiken an Gesundheitsdienstleister und Patienten.
Früher konzentrierte sich die FDA auf Funktionssicherheit Aspekte der Systeme, aber jetzt Internet-Sicherheit ist ein Thema von gleicher Bedeutung. Auch wenn Safety und Security ähnlich sind – und man könnte leicht argumentieren, dass es bei beiden um die Entwicklung vorhersehbarer Software geht – betrachtet die FDA Cybersicherheit als etwas, das besondere Aufmerksamkeit und Maßnahmen erfordert.
Die Bedeutung der Prozessvalidierung bei Medizinprodukten
Bei der Prozessvalidierung handelt es sich um die Sammlung und Auswertung von Daten, die den wissenschaftlichen Nachweis erbringen, dass ein Prozess in der Lage ist, dauerhaft ein Qualitätsprodukt zu liefern. Im Zusammenhang mit Medizinprodukten ist die Prozessvalidierung von entscheidender Bedeutung, um die Sicherheit und Wirksamkeit der Geräte über ihren gesamten Lebenszyklus hinweg zu gewährleisten.
Die Prozessvalidierung ist besonders wichtig für medizinische Geräte, die durch Software gesteuert werden, da Software komplex und schwer umfassend zu testen sein kann. Beispielsweise kann ein softwaregesteuertes medizinisches Gerät Millionen von Codezeilen umfassen, und es ist nicht möglich, jedes mögliche Szenario und jede mögliche Kombination von Ein- und Ausgängen zu testen, ohne beide zu nutzen statische Codeanalyse und dynamische Analyse.
Im Folgenden sind drei Hauptgründe aufgeführt, warum Prozessvalidierung wichtig ist.
- Qualitätssicherung. Die Prozessvalidierung trägt dazu bei, dass Medizinprodukte konsistent und zuverlässig hergestellt werden, wodurch das Fehlerrisiko verringert und ein hohes Qualitätsniveau sichergestellt wird.
- Patientensicherheit. Durch die Validierung des Herstellungsprozesses können potenzielle Risiken und Mängel identifiziert und gemindert werden, wodurch die Patientensicherheit erhöht wird.
- Einhaltung regulatorischer Anforderungen. Die Erfüllung regulatorischer Anforderungen, wie beispielsweise der Quality System Regulation (QSR) der FDA, erfordert den Einsatz validierter Prozesse. Andernfalls kann es zu Compliance-Problemen kommen.
Regulatorische Anforderungen und das FDA Guidance Document
Die in der FDA-Leitfaden zur Cybersicherheit medizinischer Geräte sollen den wachsenden Bedenken hinsichtlich der Sicherheit medizinischer Geräte in einem zunehmend vernetzten und digitalen Gesundheitsumfeld Rechnung tragen. Hier finden Sie eine Erläuterung der regulatorischen Anforderungen.
Gesetzliche Befugnis
Die FDA verfügt nun über neue gesetzliche Befugnisse im Rahmen des Food and Drug Omnibus Reform Act (FDORA), der im Dezember 2022 in Kraft trat. Dieses Gesetz ermöglicht es der FDA, bei Einreichungen von Medizinprodukten für „Cyber-Geräte“ Informationen zur Cybersicherheit zu verlangen und die Hersteller dazu zu verpflichten, konkrete Vorgaben zu machen Maßnahmen zum Nachweis hinreichender Sicherheit, dass ihre Geräte und zugehörigen Systeme „cybersicher“ sind. Die Nichteinhaltung dieser Anforderungen gilt als verbotene Handlung und wird strafrechtlich verfolgt.
Definition von „Cyber-Gerät“
Der Begriff „Cyber-Gerät“ wird im Gesetz neu definiert als ein Gerät, das vom Sponsor validierte, installierte oder autorisierte Software enthält, eine Verbindung zum Internet herstellen kann und technologische Merkmale aufweist, die für Cybersicherheitsbedrohungen anfällig sein könnten. Diese Definition hilft zu klären, welche Geräte den neuen Cybersicherheitsanforderungen unterliegen.
Einbeziehung von Cybersicherheitsinformationen in Premarket-Einreichungen
Die neuen Leitlinien schreiben vor, dass Hersteller in ihren Anträgen vor der Markteinführung bei der FDA spezifische Informationen zur Cybersicherheit angeben müssen. Zu diesen Premarket-Einreichungen können Premarket Approval (PMA), 510(k)- oder De-novo-Einreichungen gehören. Die Cybersicherheitsinformationen sind erforderlich, um sicherzustellen, dass das Gerät die im Gesetz festgelegten Cybersicherheitsanforderungen erfüllt.
Überwachung und Behebung von Cybersicherheitsschwachstellen nach der Markteinführung
Gerätehersteller müssen einen Plan zur Überwachung, Identifizierung und Behebung von Cybersicherheitsschwachstellen und Exploits nach dem Inverkehrbringen vorlegen. Dieser Plan sollte den Ansatz des Herstellers zur kontinuierlichen Überwachung und Bewältigung von Cybersicherheitsrisiken darlegen, die nach der Markteinführung des Geräts auftreten können.
Angemessene Gewährleistung der Cybersicherheit
Hersteller müssen Prozesse und Verfahren entwerfen, entwickeln und aufrechterhalten, um hinreichende Sicherheit dafür zu bieten, dass das Gerät und die zugehörigen Systeme „cybersicher“ sind. Diese Anforderung unterstreicht die Notwendigkeit eines proaktiven Ansatzes, um Sicherheit in das Design des Geräts zu integrieren und diese über den gesamten Lebenszyklus des Geräts aufrechtzuerhalten.
Software-Stückliste
Von den Herstellern wird erwartet, dass sie im Rahmen ihrer Einreichungen eine Software-Stückliste (SBOM) bereitstellen. In dieser Rechnung sollten die kommerziellen, Open-Source- und Standardsoftwarekomponenten aufgeführt sein, die im Gerät verwendet werden. Diese Informationen sind entscheidend für die Identifizierung und Behebung potenzieller Schwachstellen im Software-Stack.
Einhaltung zusätzlicher FDA-Anforderungen
Das Leitliniendokument ermächtigt die FDA außerdem, durch Vorschriften zusätzliche Anforderungen festzulegen, um nachzuweisen, dass das Gerät und die zugehörigen Systeme cybersicher sind. Hersteller müssen darauf vorbereitet sein, alle zusätzlichen Anforderungen der FDA zu erfüllen.
Verbotenes Gesetz wegen Nichteinhaltung
Ein wesentlicher Aspekt der regulatorischen Anforderungen ist die Aufnahme eines neuen gesetzlichen Verbotsgesetzes. Das bedeutet, dass die Nichteinhaltung der Cybersicherheitsanforderungen der FDA nicht nur ein Compliance-Problem, sondern auch einen Rechtsverstoß darstellt. Die Regierung ist befugt, Verstöße gegen diese Anforderungen strafrechtlich zu verfolgen oder Unterlassungsansprüche gegen nicht konforme Unternehmen geltend zu machen.
Wie Hersteller medizinischer Geräte die Anforderungen erfüllen können
Hersteller von Medizinprodukten müssen viele Anforderungen erfüllen, um die Sicherheit, Wirksamkeit und Qualität ihrer Produkte zu gewährleisten. Wir werden uns mit den wichtigsten Aspekten der Einhaltung der FDA-Vorschriften für Medizinprodukte befassen, wobei wir einen größeren Schwerpunkt auf die Berücksichtigung von Sicherheitsbedenken bei Medizinprodukten und die Entwicklung von Kontroll- und Validierungsprotokollen für Hersteller legen.
Bewältigung von Sicherheitsbedenken bei medizinischen Geräten
Um die FDA-Vorschriften einzuhalten und der Patientensicherheit Priorität einzuräumen, müssen Hersteller medizinischer Geräte Sicherheitsbedenken wirksam angehen. Hier sind einige Tipps, um Sicherheitsbedenken bei medizinischen Geräten auszuräumen:
- Standardisieren Sie Ihren Code und führen Sie eine Risikobewertung durch. Der erste Schritt zu einem medizinischen Softwaregerät ohne Sicherheitsprobleme besteht darin, die Software mit standardisierten Codierungsregeln schreiben zu lassen. Bei bereits vorhandener Software können Sie die Codebasis überprüfen und bereinigen, um sicherzustellen, dass sie akzeptablen Codierungsstandards entspricht. Hersteller medizinischer Geräte können statische Code-Analyselösungen nutzen, um den Quellcode ihrer Softwareprogramme zu testen und zu analysieren. Neben der statischen Codeanalyse können Integrationstests, Sicherheitstests, Unit-Tests, Systemtests und Usability-Tests zu weiteren Arten von Softwaretests beitragen, die zur Bereitstellung großartiger Software beitragen können. Durch die Bereinigung der Software-Codebasis und die Durchführung einer Risikoanalyse können Hersteller potenzielle Sicherheitslücken in ihren Geräten identifizieren. Dazu gehört die Bewertung von Risiken im Zusammenhang mit Datenschutzverletzungen, unbefugtem Zugriff und der Integrität der Gerätefunktionalität.
- Software-Updates und Patch-Management. Aktualisieren und patchen Sie die Gerätesoftware regelmäßig, um bekannte Schwachstellen zu beheben. Rechtzeitige Updates tragen dazu bei, Sicherheitsverletzungen zu verhindern, die möglicherweise veraltete Software ausnutzen.
- Sichere Datenspeicherung. Stellen Sie sicher, dass Patientendaten sowohl auf dem Gerät als auch während der Datenübertragung sicher gespeichert werden. Nutzen Sie sichere Datenspeicherungspraktiken und von der Industrie empfohlene Verschlüsselungsprotokolle, um die Privatsphäre der Patienten zu schützen und die Datenschutzbestimmungen einzuhalten.
- Arbeiten Sie mit Cybersicherheitsexperten zusammen. Erwägen Sie die Zusammenarbeit mit Cybersicherheitsexperten und Fachleuten, die auf die Sicherheit medizinischer Geräte spezialisiert sind. Ihr Fachwissen kann Herstellern dabei helfen, potenzielle Sicherheitsrisiken zu erkennen und zu mindern.
Designkontroll- und Validierungsprotokoll für Hersteller
Richtige Designkontrollen und Validierungsprotokolle sind wichtige Bestandteile, um sicherzustellen, dass medizinische Geräte die regulatorischen Anforderungen der FDA erfüllen. Diese Kontrollen helfen Herstellern, sichere und wirksame Geräte zu entwickeln, die den Patienten die beabsichtigten Vorteile bieten.
Hier finden Sie wichtige Überlegungen zur Implementierung von Designkontrollen und Validierungsprotokollen.
- Design- und Entwicklungsplanung. Hersteller sollten einen klaren Design- und Entwicklungsplan erstellen, der den Umfang, die Ziele und die Erwartungen für das Medizinprodukt umreißt. Dieser Plan sollte Risikomanagement, Usability-Studien und die Einhaltung gesetzlicher Vorschriften umfassen.
- Entwerfen Sie Ein- und Ausgänge. Definieren Sie Designeingaben, die die Anforderungen des Geräts spezifizieren, und Designausgaben, die die Eigenschaften des Geräts beschreiben. Stellen Sie sicher, dass die Designeingaben den Benutzerbedürfnissen und gesetzlichen Anforderungen entsprechen.
- Verifizierung und Validierung. Implementieren Sie strenge Verifizierungs- und Validierungsprozesse, um zu bestätigen, dass das Gerät seinen Designanforderungen und seinem Verwendungszweck entspricht. Dazu gehört die Prüfung und Bewertung der Leistung und Sicherheit des Geräts.
- Designhistoriedatei (DHF). Pflegen Sie ein umfassendes DHF, das alle Design- und Entwicklungsaktivitäten aufzeichnet, einschließlich der Dokumentation von Designkontrollen, Testergebnissen und Designänderungen.
- Zulassungsanträge. Das Einreichen genauer und vollständiger Premarket-Benachrichtigungen (510(k)), De-Novo-Anträge oder Premarket-Zulassungsanträge (PMA) ist für die Erlangung der FDA-Freigabe oder -Zulassung für Ihre Medizinprodukte von entscheidender Bedeutung. Diese Einreichungen liefern der FDA die notwendigen Informationen über die Sicherheit und Wirksamkeit Ihres Geräts und helfen ihr bei der Beurteilung seiner Markttauglichkeit. Wenn Sie diese Einreichungen nicht korrekt ausfüllen, kann es zu Verzögerungen oder sogar zur Ablehnung Ihres Produkts kommen.
- Postmarket Surveillance (PMS). Die Einrichtung eines PMS-Systems ist für die Überwachung und Meldung unerwünschter Ereignisse, Beschwerden und Gerätefehlfunktionen von entscheidender Bedeutung. Die Einhaltung der PMS-Anforderungen hilft, Sicherheitsprobleme zu erkennen und Korrekturmaßnahmen schnell umzusetzen. Es stellt sicher, dass Hersteller die Leistung ihrer Geräte aktiv überwachen und alle Probleme beheben, die sich aus der Nutzung ergeben.
Tools und Methoden zur FDA-Konformität
Hersteller können verschiedene Tools und Methoden einsetzen, um die Einhaltung der FDA-Vorschriften für Medizinprodukte zu erreichen und aufrechtzuerhalten. Im Folgenden werden wesentliche Tools und Methoden besprochen, die bei der Verfolgung der FDA-Konformität hilfreich sein können.
Statische Analyse zur Erfüllung der FDA-Anforderungen
Die statische Analyse spielt eine entscheidende Rolle dabei, Herstellern medizinischer Geräte dabei zu helfen, die strengen Anforderungen der FDA zu erfüllen.
Hier sind 5 Gründe, statische Analysen zur Erfüllung der FDA-Anforderungen einzusetzen.
- Frühzeitige Problemerkennung. Statische Analysewerkzeuge kann potenzielle Probleme wie Codierungsfehler, Sicherheitslücken und Designfehler bereits in einem frühen Stadium des Entwicklungsprozesses erkennen. Diese Früherkennung ermöglicht zeitnahe Korrekturen und verringert so das Risiko kostspieliger und zeitaufwändiger Änderungen später im Entwicklungszyklus. Empfohlene Codierungsstandards für C und C++ sind MISRA C / C ++ und CERT C/C++.
- Verbesserte Softwarequalität. Hochwertige Software ist für die Sicherheit und Wirksamkeit medizinischer Geräte unerlässlich. Die statische Analyse hilft Entwicklern dabei, saubereren, zuverlässigeren und sichereren Code zu erstellen. Dies erleichtert nicht nur die FDA-Konformität, sondern verringert auch das Risiko von Problemen und Sicherheitsbedenken nach der Markteinführung.
- Einhaltung der Cybersicherheit. Angesichts der zunehmenden Bedeutung der Cybersicherheit für medizinische Geräte legt die FDA großen Wert darauf. Statische Analysetools können Sicherheitsschwächen und Schwachstellen in der Software identifizieren und so die Übereinstimmung mit den FDA-Richtlinien zur Cybersicherheit sicherstellen.
- Regulatorische Dokumentation. Durch die statische Analyse können umfassende Dokumentationen und Berichte bereitgestellt werden, die die gründliche Prüfung der Software und die zur Behebung festgestellter Probleme ergriffenen Schritte veranschaulichen. Diese Dokumentation ist für FDA-Einreichungen und -Inspektionen unerlässlich.
- Optimierte Validierung. Durch die frühzeitige Identifizierung und Behebung potenzieller Probleme kann die statische Analyse den Validierungsprozess beschleunigen, was einen wichtigen Schritt zur Einhaltung der FDA-Konformität darstellt. Dies trägt dazu bei, Verzögerungen bei der Markteinführung medizinischer Geräte zu reduzieren.
So stellen Sie ein ordnungsgemäßes Risikomanagement sicher
Angesichts der Rolle des Risikomanagements bei der Erreichung der allgemeinen Ziele oder Anforderungen der FDA wollen wir uns damit befassen, wie Sie ein angemessenes Risikomanagement erreichen können.
- Risikoabschätzung. Die Risikobewertung bei der Herstellung von Medizinprodukten ist der erste Schritt zur Identifizierung und zum Verständnis potenzieller Gefahren, die mit einem Medizinprodukt verbunden sind. Sein Zweck besteht darin, das Design, den Verwendungszweck, die Biokompatibilität und andere Risikofaktoren des Geräts systematisch zu bewerten, um potenzielle Risiken zu ermitteln. Bei einer Risikobewertung wird in der Regel ein funktionsübergreifendes Team mit unterschiedlichem Fachwissen gebildet, darunter Ingenieure, Kliniker und Regulierungsspezialisten.
- Risikoanalyse. Ziel der Risikoanalyse ist es, die identifizierten Risiken zu bewerten und zu priorisieren, indem eine Risikobewertung basierend auf der Eintrittswahrscheinlichkeit und der Schwere des potenziellen Schadens zugewiesen wird. Bei der Risikoanalyse wird jedes Risiko quantifiziert, indem seine Wahrscheinlichkeit und mögliche Auswirkung bewertet werden. Abhängig von Ihrem Unternehmen kann eine Risikomatrix oder ein anderes Bewertungssystem zur Priorisierung von Risiken verwendet werden. Hochriskante Gegenstände erhalten bei der weiteren Bearbeitung Vorrang.
- Risikominderung. Zur Risikominderung gehört die Entwicklung von Strategien zur Reduzierung oder Kontrolle identifizierter Risiken auf ein akzeptables Maß. Ziel ist es, die Sicherheit und Wirksamkeit des Medizinprodukts zu verbessern. Abhilfemaßnahmen können verschiedene Formen annehmen, darunter Designänderungen, Sicherheitsmerkmale, bessere Materialien, verbesserte Benutzerschulungen oder verbesserte Gebrauchsanweisungen. Ziel ist es, die identifizierten Risiken so anzugehen, dass ihre Auswirkungen minimiert werden.
- Dokumentation. Eine ordnungsgemäße Dokumentation ist für Transparenz, Rechenschaftspflicht und Einhaltung gesetzlicher Vorschriften von entscheidender Bedeutung. Es bietet eine Aufzeichnung des Risikomanagementprozesses und hilft, die Sorgfaltspflicht gegenüber den Aufsichtsbehörden nachzuweisen. Alle Aspekte des Risikomanagementprozesses, einschließlich Risikobewertungen, -analysen und -minderungsplänen, sollten sorgfältig dokumentiert werden. Diese Dokumentation wird während des gesamten Produktlebenszyklus gepflegt.
- Ständige Verbesserung. Der Risikomanagementprozess sollte ein dynamisches und sich weiterentwickelndes System sein. Kontinuierliche Verbesserung stellt sicher, dass Ihre Risikomanagementpraktiken bei der Bewältigung neuer Risiken und sich ändernder Marktbedingungen wirksam bleiben. Regelmäßige Überprüfungen und Aktualisierungen werden durchgeführt, um Erkenntnisse aus früheren Projekten zu berücksichtigen und sich an die sich entwickelnde Technologie und Vorschriften anzupassen. Dies kann die Überprüfung und Überarbeitung von Risikobewertungen, -analysen und -minderungsplänen beinhalten.
Wie die statische Analyse für C/C++ beim Risikomanagement helfen kann
Viele unserer Kunden von Medizinprodukten haben von Anfang an Erfolge bei der Einführung erzielt statische Analyse für C / C ++ indem Sie die folgenden Schritte ausführen:
- Überprüfen Sie bestehende Richtlinien in der Organisation. Auch wenn sie für die manuelle Durchsetzung konzipiert sind, ordnen Sie sie so weit wie möglich den Prüfern zu, die mit dem statischen Analysetool bereitgestellt werden. Ein ausgereiftes statisches Analysetool wird wahrscheinlich die meisten davon abdecken. Sie können erwägen, benutzerdefinierte Prüfer für die verbleibenden Richtlinien zu erstellen, die nicht sofort statischen Analyseprüfern zugeordnet werden konnten.
- Sehen Sie sich die gängigen Codierungsstandards an, insbesondere solche, die unter Berücksichtigung von Sicherheitsaspekten erstellt wurden. Wählen Sie eine Untergruppe von Richtlinien aus, die Ihr Team befolgen soll. Bei der Auswahl der Richtlinien ist es sinnvoll, sich an der Kategorisierung aus den Standards zu orientieren und die Richtlinien auszuwählen, die als die wichtigsten eingestuft werden. Beispielsweise möchten Sie in CERT wahrscheinlich mit L1-Richtlinien beginnen und in MISRA C 2023 verbindliche Richtlinien überprüfen.
- Definieren Sie die Konfiguration des statischen Analysetools und beziehen Sie Ihre organisationsspezifischen Richtlinien und ausgewählte Richtlinien wie CERT ein. Aktivieren Sie nicht alle Checker gleichzeitig. Beginnen Sie stattdessen mit einer kleinen Teilmenge, um zu vermeiden, dass Entwickler mit Verstößen überschwemmt werden.
- Stellen Sie sicher, dass Ihre Entwickler ihren Code sofort nach der Erstellung scannen können. Sinnvoll ist auch die Einbeziehung der statischen Analyse in den CI/CD-Prozess.
- Während die Entwickler Fortschritte machen und den Quellcode bereinigen, stellen Sie sicher, dass Sie nach und nach weitere Prüfer aus Ihrer Liste aktivieren. Am Ende des Tages möchten Sie den Nachweis erbringen, dass Sie alle von Ihnen ausgewählten Richtlinien einhalten und nicht, dass Sie mittendrin aufgehört haben. Um sich im Prozess besser zu orientieren und Ihren aktuellen Fortschritt zu verstehen, können Sie ein zentrales Berichtssystem einsetzen, das dabei hilft, die Testdaten zusammenzufassen und die Arbeit der Entwickler zu überwachen. Sehen Sie sich unten ein Beispiel dafür an:
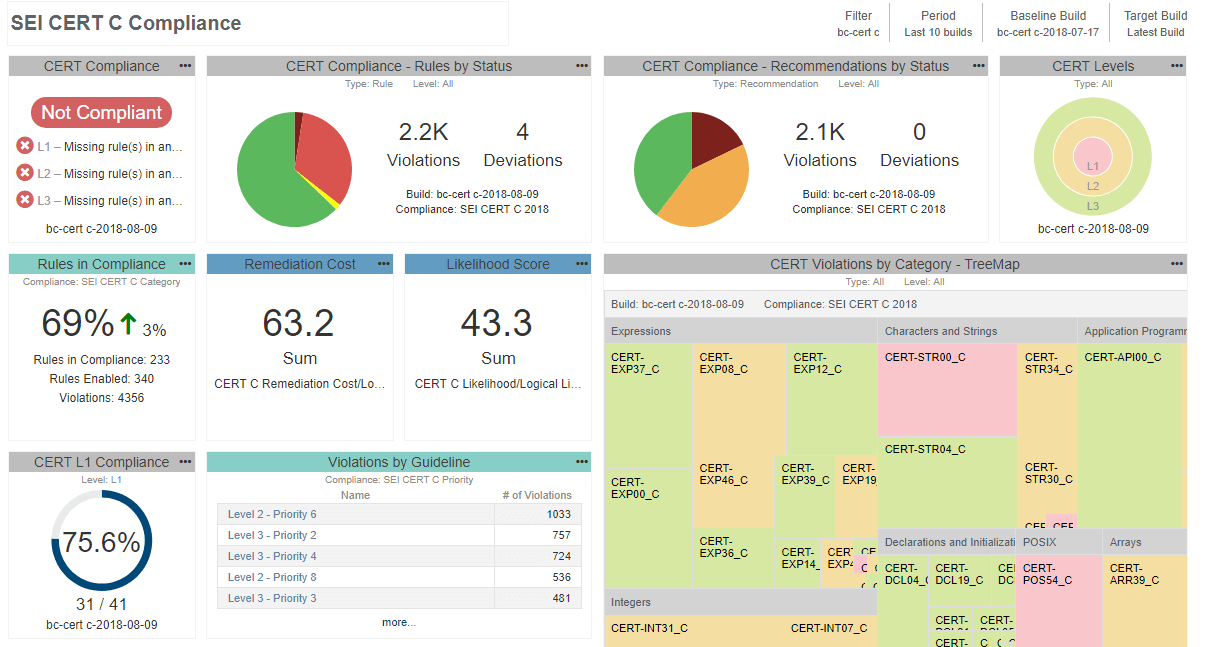
Da statische Analyseberichte Teil des Qualitätsmanagementsystems werden, können Sie nicht jedes beliebige Tool verwenden. Die FDA verlangt, dass alle bei der Entwicklung und Verifizierung der Software verwendeten Tools für den vorgesehenen Verwendungszweck validiert werden. Es gibt verschiedene Möglichkeiten, die Eignung des Tools für den Einsatz in sicherheitskritischen Entwicklungen nachzuweisen. Abhängig vom Risiko des Geräts kann es so einfach sein, ein Konformitätszertifikat wiederzuverwenden oder den längeren Prozess abzuschließen Werkzeugqualifikation.
Verwendung einer TÜV SÜD-Zertifizierung
Für den Endbenutzer ist es am bequemsten, die Gutschrift für die vom Werkzeughersteller geleistete Arbeit zu erhalten und die Zertifizierung, die von einer externen Zertifizierungsorganisation wie TÜV SÜD für das Testwerkzeug erteilt wurde, wiederzuverwenden. Parasoft C / C ++ test, zum Beispiel, ist mit abgedeckt eine TÜV SÜD-Zertifizierung die wiederverwendet werden können, um die Eignung für die Entwicklung von Software gemäß medizinischen Standards wie IEC 62304 nachzuweisen.
Durchführen der Werkzeugqualifizierung
Für Hochrisikogeräte wie Klasse C müssen Sie das Tool möglicherweise intern in Ihrer Entwicklungsumgebung validieren. Ziel ist es, den Nachweis zu erbringen, dass das Tool gemäß seinen in der Entwicklungsumgebung des Projekts gesammelten betrieblichen Anforderungen funktioniert. Dies ist ein sehr langwieriger und zeitaufwändiger Prozess.
Am besten ist es, wenn Ihr Tool-Anbieter Sie bei diesem Vorhaben unterstützen und ein spezielles Tool-Qualifizierungskit bereitstellen kann, das gut gestaltete Testfälle und das Automatisierungs-Framework enthält, um diese in der Entwicklungsumgebung des Projekts auszuführen und automatisch die Dokumentation zu generieren, die als dienen kann der Beweis für die Werkzeugvalidierung. Auch hier bietet Parasofts Flaggschiffprodukt C/C++test eine automatisiertes Werkzeugqualifizierungskit.
Fallstudien und reale Anwendungen
Wir verfügen über eine Fülle von Fallstudien zu realen Anwendungen der statischen Analysetools von Parasoft. Aber schauen wir uns zwei unterschiedliche Erfolgsgeschichten an, die veranschaulichen, wie unsere Lösung für die statische Analyse eine wichtige Rolle dabei gespielt hat, Herstellern medizinischer Geräte dabei zu helfen, ihre einzigartigen Herausforderungen zu meistern und die FDA-Anforderungen zu erfüllen.
Erfolgsgeschichten von Herstellern medizinischer Geräte
Die folgenden Fallstudien veranschaulichen, wie die Softwaretestlösungen von Parasoft maßgeblich dazu beigetragen haben, Hersteller medizinischer Geräte bei der Bewältigung ihrer einzigartigen Herausforderungen und beim Erreichen ihrer Ziele zu unterstützen. Inovytec und Smiths Medical verzeichneten durch ihre Zusammenarbeit mit Parasoft beide bemerkenswerte Verbesserungen bei der Codequalität, Compliance und Testeffizienz.
Inovytecs Weg zur FDA 510(k)-Zertifizierung
Inovytec, ein Unternehmen, das sich auf die Herstellung medizinischer Geräte spezialisiert hat, hat sich zum Ziel gesetzt, die FDA 510(k)-Zertifizierung für sein Beatmungsgerät Ventway Sparrow zu erhalten. Ihre Herausforderung bestand darin, sauberen Code zu liefern und gleichzeitig die FDA-Vorschriften einzuhalten. Die statische Codeanalyselösung C/C++ von Parasoft kam ihnen zu Hilfe.
Das Softwareentwicklungsteam von Inovytec hat den Parasoft C/C++-Test angepasst, um ihn an die strengen FDA-Anforderungen anzupassen. Jedes Mal, wenn sie sich auf die Veröffentlichung einer neuen Softwareversion vorbereiteten, stellten sie sicher, dass die statische Analyse von Parasoft so konfiguriert war, dass sie gemäß den Definitionen der FDA-Verordnung ausgeführt wurde. Das Ergebnis war nicht nur eine verbesserte Codequalität, sondern auch ein durchschlagender Erfolg bei der 100-prozentigen Erfüllung der FDA 510(k)-Zertifizierungsregeln und -richtlinien. Parasoft erwies sich bei Inovytec als bevorzugte Testlösung und die Zusammenarbeit mit ESL, einem Vertriebshändler für Parasoft-Produkte in Israel, bot bei Bedarf wichtige Unterstützung und Fachwissen.
Einführung der testgetriebenen Entwicklung (TDD) durch Smiths Medical
Smiths Medical, ein renommierter Hersteller von medizinischen Spezialgeräten, sah sich bei seinem Bestreben, qualitativ hochwertige, sicherheitskritische Geräte zu entwickeln, mit einer Reihe von Herausforderungen konfrontiert Software für medizinische Geräte. Automatisierte Tests spielen eine entscheidende Rolle in der Teststrategie von Smiths Medical.
Frühere Bemühungen, Tools zu integrieren, waren nicht von vollem Erfolg. Das Entwicklungsteam suchte nach einer Lösung, die seinen gesamten Testprozess verbessern könnte, indem es eine neue Perspektive einnimmt, die sich auf Unit-Tests und testgetriebene Entwicklung (TDD) konzentriert, eine Methodik, die Design, Tests und Codeentwicklung integriert. Sie brauchten ein Tool, das in ihre Testpipeline passt und ihre gesamte Entwicklungskultur verbessert. Parasoft C/C++test erwies sich als die Antwort auf ihre Herausforderungen.
Das Softwareteam erzielte nicht nur eine erfolgreiche TDD-Einführung, sondern profitierte auch von einer verbesserten Teststabilität, einer verbesserten Codeabdeckung und einem optimierten Tool-Qualifizierungsprozess, was für sicherheitskritische Anwendungen von entscheidender Bedeutung war. Mit Parasoft war Smiths Medical in der Lage, seine Entwicklungsprozesse zu transformieren, Tests zu einem integralen Bestandteil seiner Software-Pipeline zu machen und letztendlich die Lieferung sicherer, qualitativ hochwertiger medizinischer Geräte sicherzustellen.
Herausforderungen bei der FDA-Validierung meistern
Der FDA-Validierungsprozess kann für Hersteller medizinischer Geräte komplex und herausfordernd sein. Es gibt jedoch eine Reihe von Maßnahmen, die Hersteller ergreifen können, um diese Herausforderungen zu meistern.
- Früh anfangen. Der FDA-Validierungsprozess sollte früh im Produktentwicklungszyklus beginnen. Dies gibt den Herstellern mehr Zeit für die Planung und Durchführung des Validierungsprozesses und hilft, potenzielle Probleme frühzeitig zu erkennen und zu beheben.
- Verwenden Sie einen risikobasierten Ansatz. Der FDA-Validierungsprozess sollte auf einer Risikobewertung des Geräts basieren. Hersteller sollten ihre Validierungsbemühungen auf die kritischsten Komponenten und Funktionen des Geräts konzentrieren.
- Setzen Sie qualifiziertes Personal ein. Der FDA-Validierungsprozess sollte von qualifiziertem Personal durchgeführt werden, das Erfahrung mit FDA-Vorschriften und Best Practices für die Validierung hat.
- Dokumentieren Sie den Validierungsprozess. Hersteller sollten den FDA-Validierungsprozess gründlich dokumentieren. Dadurch können Hersteller gegenüber der FDA nachweisen, dass sie ihr Gerät gemäß den FDA-Vorschriften validiert haben.
Nächste Schritte zum Navigieren in den FDA-Validierungsrichtlinien
Die FDA stellt Herstellern medizinischer Geräte wichtige Validierungsrichtlinien zur Verfügung, um Produktqualität und Patientensicherheit sicherzustellen. Da sich Vorschriften und Richtlinien weiterentwickeln, ist es von entscheidender Bedeutung, über diese Änderungen auf dem Laufenden zu bleiben und die zukünftige Einhaltung zu planen. Medizingerätehersteller können dies auf zweierlei Weise erreichen: indem sie sich auf zukünftige FDA-Richtlinien vorbereiten und diese einhalten.
Vorbereitung auf zukünftige FDA-Richtlinien
Die FDA aktualisiert und überarbeitet ständig ihre Leitliniendokumente für Hersteller von Medizinprodukten. Um sich auf zukünftige FDA-Richtlinien vorzubereiten, sollten Hersteller medizinischer Geräte:
- Überwachen Sie die Website der FDA auf Aktualisierungen der Leitliniendokumente. Die FDA veröffentlicht Aktualisierungen ihrer Leitliniendokumente auf ihrer Website. Hersteller sollten regelmäßig auf der Website der FDA nach Aktualisierungen der Leitfäden suchen, die für ihre Produkte relevant sind.
- Nehmen Sie an Workshops und Webinaren der FDA teil. Die FDA veranstaltet regelmäßig Workshops und Webinare zu verschiedenen Themen im Zusammenhang mit der Regulierung von Medizinprodukten. Hersteller sollten an diesen Veranstaltungen teilnehmen, um über die neuesten Überlegungen der FDA zur Regulierung von Medizinprodukten auf dem Laufenden zu bleiben.
- Vernetzen Sie sich mit anderen Medizingeräteherstellern. Hersteller können sich mit anderen Medizingeräteherstellern vernetzen, um Informationen und Best Practices zur Einhaltung der FDA-Vorschriften auszutauschen.
Kontinuierliche Compliance sicherstellen
Hersteller medizinischer Geräte müssen die kontinuierliche Einhaltung der FDA-Vorschriften sicherstellen. Um dies zu erreichen, sollten Hersteller:
- Ein Qualitätssystem einrichten und aufrechterhalten. Ein Qualitätssystem ist eine Reihe von Verfahren und Prozessen, mit denen Hersteller die Sicherheit und Wirksamkeit ihrer Geräte gewährleisten. Hersteller sollten ein Qualitätssystem einrichten und aufrechterhalten, das den Anforderungen der Quality System Regulation der FDA entspricht.
- Führen Sie regelmäßige Audits ihres Qualitätssystems durch. Hersteller sollten ihr Qualitätssystem regelmäßig überprüfen, um etwaige Verstöße zu identifizieren und zu beheben.
- Senden Sie MDR-Berichte an die FDA. Hersteller sind verpflichtet, bei der FDA Medizingeräteberichte (Medical Device Reports, MDR) für unerwünschte Ereignisse einzureichen, die bei ihren Geräten auftreten. Durch die Übermittlung von MDR-Berichten an die FDA können Hersteller der FDA dabei helfen, Sicherheitsbedenken hinsichtlich ihrer Geräte zu erkennen und zu beheben.
Im Laufe der Zeit hat sich das Erreichen der FDA-Konformität als recht streng und zeitaufwändig erwiesen. Mit statischen und dynamischen Analysetools können diese Herausforderungen jedoch gemeistert werden. Statische Analysetools bieten erweiterte Funktionen zur statischen Codeanalyse, die dabei helfen, Probleme in Software und Code frühzeitig im Entwicklungsprozess zu erkennen und zu beheben.
Die Einführung einer statischen Analyse ist ein besonderer Aufwand, der Entwicklerzeit und -kosten erfordert. Aber es ist eine bewährte Methode, Ihr System gegen böswillige Angriffe abzusichern. Durch den Einsatz einer statischen Analyse mit gut durchdachten Sicherheitsrichtlinien können Sie Systeme aufbauen, die unvorhergesehenen zukünftigen Angriffen standhalten können.
IEC 62304-Softwarekonformität in der Medizinbranche
„MISRA“, „MISRA C“ und das Dreieckslogo sind eingetragene Marken von The MISRA Consortium Limited. ©The MISRA Consortium Limited, 2021. Alle Rechte vorbehalten.
Weiterführende Inhalte

Whitepaper (ENG)


